Germline short variant discovery (SNPs + Indels) and interpretation
Design document
Table of contents
Introduction
DNA sequence data is received from the sequencing facility in FASTQ format. It is generally considered that approximately 20-40% of rare genetic disease may be detectable by DNA germline short variant discovery and variant interpretation Specifically, this is tailored to single nucleotide variants (SNVs) and short insertion/deletions (INDELs) in protein coding genes and splice sites.
To produce our final output (e.g. clinical grade reports, downstream statistical analysis), we must process the data as follows:
- QC for pre-processing
- Decide on protocol, alignment build, threshold criteria
- Annotate known effect
- Interpret impact
- Report clinical grade genetics
- Cohort analysis for variant association
- Any additional custom outputs
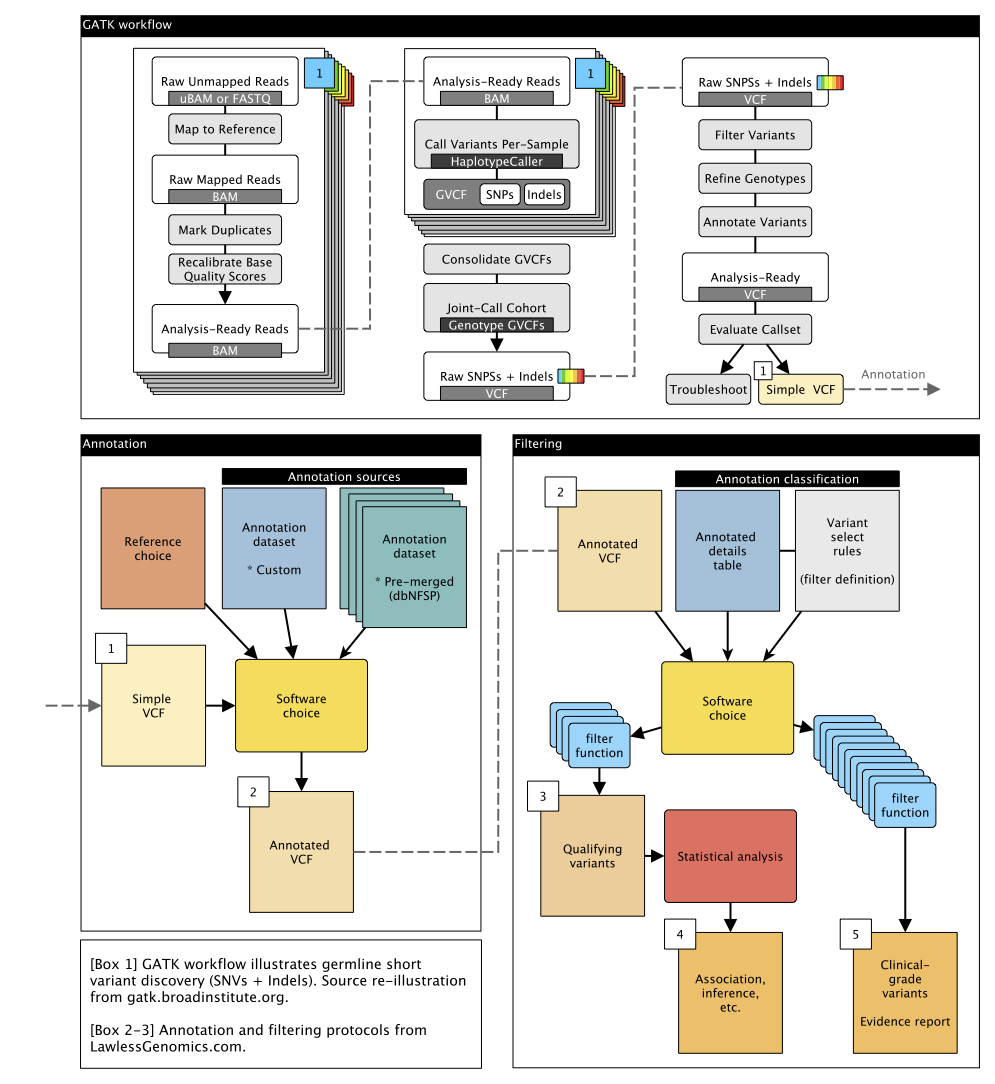 Figure: Summary of DNA germline short variant discovery pipeline plan.
Figure: Summary of DNA germline short variant discovery pipeline plan.
Summary of requirements
Quality control
We use a number of tools such as github.com/OpenGene/fastp for comprehensive quality profiling for both before and after filtering data.
Alignment
- We use BWA for alignment with GRCh38.
- Our reference genome build is GRCh38 from following source:
ftp://ftp.ncbi.nlm.nih.gov/genomes/all/GCA/000/001/405/GCA_000001405.15_GRCh38/seqs_for_alignment_pipelines.ucsc_ids/GCA_000001405.15_GRCh38_no_alt_analysis_set.fna.gz
This genome build is currently considered the best for our application. You can read more about it on Illumina’s website and Heng Li’s website.
- HPC requirements
- Array of jobs - 1 task per fastq file
- 8 cpu per task
- 6 hours per task
- 12G memory per task
- Example testing: requires 2hr 15min for a 5.3Gb fastq pair
Variant calling
We will implement the genome analysis tool kit GATK best practices workflow for germline short variant discovery (open source licence here). This workflow will be designed to operate on a set of samples constituting a study cohort; specifically, a set of per-sample BAM files that have been pre-processed as described in the GATK Best Practices for data pre-processing.
The following pages go into details:
- GATK Duplicates
- GATK BQSR
- GATK Haplotype caller
- GATK Genomic db import
- GATK Genotyping gVCFs
- GATK VQSR
- GATK Genotype refine
- Pre-annotation processing
- Pre-annotation MAF
Annotation
Variant annotation is a critical step in clinical and statistical genetics. Popular tools for applying annotation data to VCF format genetic data include:
- Variant Effect Predictor (VEP) link: VEP
- NIRVANA link: NIRVANA
- ANNOVAR link: ANNOVAR
We are using VEP with Conda, but we are likely to test additional methods (licence). Additionally, these tools must be paired with a set of data sources containing the annotation information which will be applied to each variant.
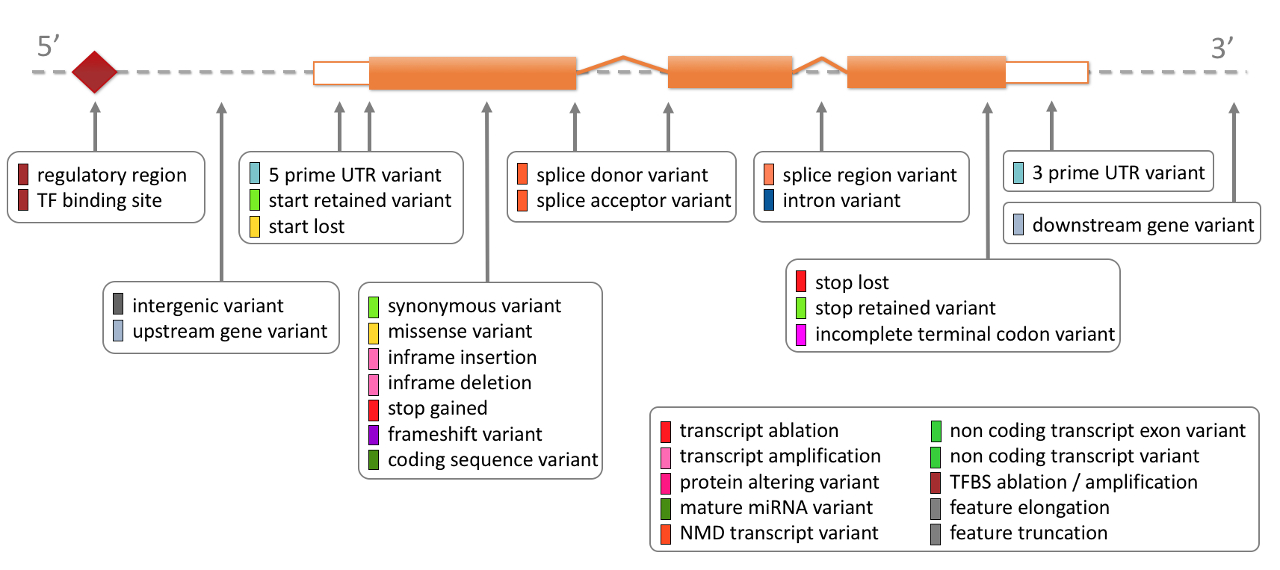
The variant consequence may be one of the defining criteria by which variants can be included in analysis since they are interpretable or of ostensibly known significance.
The consequences provided by VEP can provide a simple reference example to understand its function. For example, HIGH impact variants might be a likely consequence for identifying candidates disease-causing: Ensembl Variation - Calculated variant consequences.\
You may have observed cases in literature where clinical research reporting relied on variant effect consequence alone for known disease-related genes, but this practice is likely to introduce spurious results. It is important to use established criteria for selecting consequences of interest combined with additional filtering methods to define evidence thresholds. See the ACMG interpretation standards for examples.
Interpretation
We will perform a range of interpretation steps for:
- Generalised single-case clinical variant classification
- Cohort-level classification
For example, we will perform interpretation of variants by ACMG standards and guidelines. Extensive annotation is applied during our genomics analysis. Interpretation of genetic determinants of disease is based on many evidence sources. One important source of interpretation comes from the Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology [richards2015standards], full text at doi: 10.1038/gim.2015.30.
Check the tables linked here: temporary link. These are provided as they appear in the initial steps of our filtering protocol for the addition of ACMG-standardised labels to candidate causal variants.
- Criteria for classifications
- Caveats implementing filters
Implementing the guidelines for interpretation of annotation requires multiple programmatic steps. The number of individual caveat checks indicate the number of bioinformatic filter functions used. Unnumbered caveat checks indicate that only a single filter function is required during reference to annotation databases. However, each function depends on reference to either one or several evidence source databases (approximately 160 sources).
For reference, alternative public implementations of ACMG guidelines can be found in [li2017intervar] and [xavier2019tapes]; please note these tools have not implemented here nor is any assertion of their quality offered. Examples of effective variant filtering and expected candidate variant yield in studies of rare human disease are provided by [pedersen2021effective].
We plan to use our tools built for these requirements which are currently in review:
- ACMGuru for automated clinical genetics evidence interpretation.
- ProteoMCLustR for unbiased whole-genome pathway clustering;
- SkatRbrain for statistical sequence kernel association testing with variant collapse.
- UntangleR for pathway visualisation.
- AutoDestructR for protein structure variant mapping. All tools were designed for modular automated high-performance computing.
Table of contents
- GATK Duplicates
- GATK BQSR
- GATK Haplotype caller
- GATK Genomic db import
- GATK Genotyping gVCFs
- GATK Genotype refine
- GATK VQSR
- Pre-annotation processing
- Pre-annotation MAF